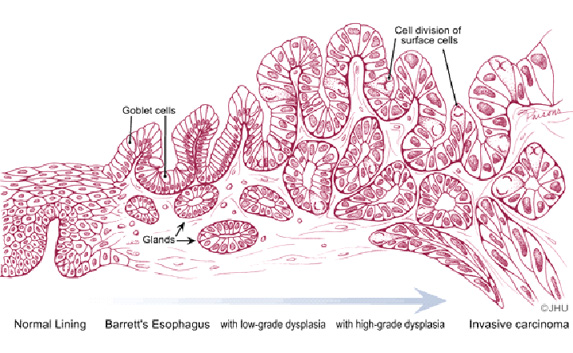
Adenocarcinoma in
Barrett esophagus develops in a sequence of changes from nondysplastic
or metaplastic columnar epithelium, through low-grade and then
high-grade dysplasia and ultimately adenocarcinoma (7). Thus critical
to the pathologic evaluation of patients with Barrett mucosa is the
degree of dysplasia, the presumed precursor of malignancy, in columnar
epithelium with intestinal metaplasia. Dysplasia is recognized by the
presence of cytologic and architectural abnormalities in the columnar
epithelium and can be classified as low-grade or high-grade, with the
predominant distinction being a basal orientation of all nuclei in
low-grade dysplasia versus nuclei consistently reaching the apex of
epithelial cells in high-grade dysplasia. Approximately 50% of patients
with high-grade dysplasia have adjacent adenocarcinoma (8). The
molecular pathogenesis of Barrett's esophagus and esophageal
adenocarcinoma has been shown to include the accumulation of multiple
genetic alterations over time. In Barrett’s esophagus, loss of
heterozygosity of such tumor suppressor genes as p53, the adenomatous
polyposis coli gene (APC), the gene deleted in colorectal cancer (DCC)
and MTS1 (p16) has been demonstrated to correlate with progression from
metaplasia to dysplasia to cancer (9).
There are three primary options once Barrett
esophagus has been diagnosed. Patients can undergo aggressive
surveillance endoscopy using the Seattle protocol (four quadrant
biopsies using jumbo biopsy forceps at 1 cm intervals and biopsy of any
mucosal irregularity with a therapeutic endoscope) at 3 month intervals
until cancer is identified, or esophagectomy or ablative therapy can be
performed. Continued surveillance using the Seattle protocol is largely
reserved for poor surgical candidates. Ablation using photodynamic
therapy (PDT) is a welcome alternative to esophagectomy for most
patients, because esophagectomy is a highly morbid surgery, even in
expert centers, with a mortality approaching 5% (10).