|
W.
Arap, E. Ruoslahti,
"Targeting the prostate for
destruction through a
vascular address"
Current surgical therapies
to remove prostate cancer
are associated with side
effects such as incontinence
and impotence. This
group is developing a
strategy to provide a less
traumatic treatment to a
prostate cancer patient by
specifically targeting the
prostate. The
selection of a homing
peptide was carried out
through phage screening as
documented in the Phage
Selection Process on this
website. The screening
found that phages expressing
two peptides selectively
homed to the prostate:
SMSIARL and VSFLEYR (single
letter amino acid codes) at
a selectively of 15 times
and 10 times more than a
control phage, respectively.
The study proceeded to use
SMSIARL to conduct in
vivo tests to target the
prostate of rats. The
SMSIARL peptide was isolated
and cloned into a T7 phage
and it showed similar
selectivity to the prostate.
Figure 1 shows the
selectivity of the SMSIARL
peptide seen during the
phage screening process.
The SMSIARL is believed to
target the vasculature of
the prostate.
Immunohistochemical staining
in Figure 2 shows that the
phage localizes around
prostate vasculature but not
around that of the brain.
The next part of this study
evaluated the ability of the
SMSIARL peptide to deliver a
biologically active drug.
p(KLAKLAK)2 is a
drug that kills bacteria but
is nontoxic to eukaryotic
cells since it cannot cross
the eukaryotic membrane.
When the SMSIARL peptide is
conjugated to p(KLAKLAK)2
through a G-G linker, it is
competent at destroying
prostate tissue.
Figure 3 shows light
microscopy histology of
mouse prostate being
destroyed by the conjugated
drug. When the two
peptides are uncoupled and
injected, the prostate does
not show any destruction
which suggests that the
SMSIARL peptide can be
coupled with other drugs to
selectively kill prostate
tumors in future studies.
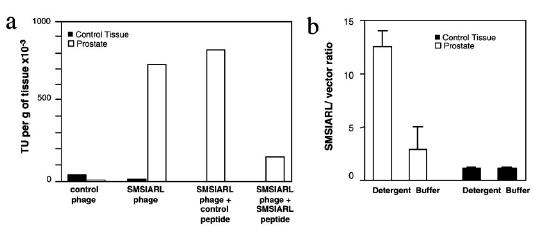
Fig. 1:
(a) Selectivity of SMSIARL
phage for the prostate.
Injection of the SMSIARL
along with the SMSIARL phage
effectively inhibited the
binding to the prostate
compared to a control
peptide. This is
perhaps the result of
competitive binding.
(b) Prostate tissue and brain
tissue (control) were tested
for incorporation of the T7
phage cloned with the
SMSIARL peptide. Phage
was extracted with either a
PBS buffer or a lysis
detergent. PCR was run
on the extracted phage
colonies to screen for the
SMSIARL sequence and in both
conditions, incorporation
was higher in the prostate
than in the brain.
This high amount recovered
in the lysis detergent
suggest that much of the
phage incorporates into the
prostate cells.
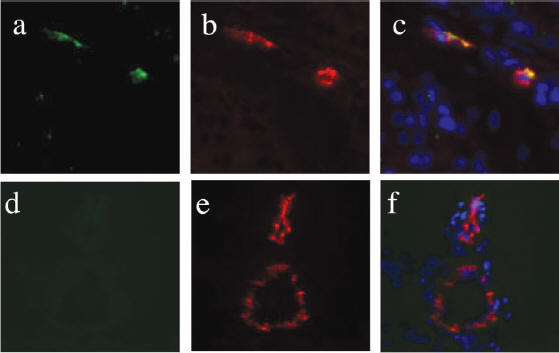
Fig. 2: (a-c) are
representative staining of
the prostate. (d-f) are
staining of brain tissue.
(a & d) are stained with an
antibody against the T7
phage and visualized with
FITC which shows green as
positive. CD31, a
marker of vascular
endothelial cells, was
targeted by an antibody and
is shown as a positive red (b,
c, e, f).
DAPI was used to stain for
nucleus (c & f) for
comparison. Similar
tests were done for kidney,
spleen, and lung tissue;
data not shown).
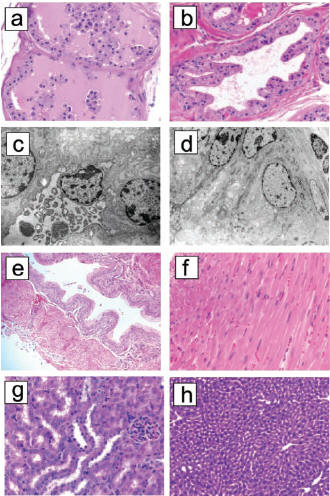
Fig. 3: (a)
Mouse prostate cross section after delivery of conjugated
SMSIARL-p(KLAKLAK)2. Massive grandular destruction
with nearly complete shedding of epithelial cells into lumen. (b)
shows the normal prostate after injection of SMSIARL and p(KLAKLAK)2
not conjugated. (c) shows the necrosis of single epithelial
cell from the conjugated group and (d) is that of the unconjugated
group. (e) is a cross section of bladder tissue from the conjugate group
which shows no damage. The same goes for heart (f), kidney (g),
and liver (h).
M.
Akerman, S. Bhatia, E.
Ruoslahti, "Nanocrystal
targeting in vivo"
This study used peptides
that homed to lung
endothelial (LE), tumor
vasculature, and human
breast carcinoma MDA-MD-435
cells. The peptides
are named GFE, F3, and LyP-1
respectively. These
peptides were adsorbed to
the surface of quantum dots
(Qdots) through a thiol-exchange
reaction. Qdots have
the unique ability to
luminesce when excited at a
certain wavelength of light.
The Qdots coated with F3
showed that the F3 tended to
aggregate, due to the
interactions of ionic
residues, and was
ineffective as a homing
device. To compensate
for this, polyethylene
glycol (PEG) was co-adsorbed
onto the Qdots to space out
the F3. Figure 1 shows
a variety of graphics which
show the preferential
binding of the three
peptides to their respective
targets and their
non-binding to other cells.
The study also found that
coating the surface of the
Qdot with PEG alongside the
peptide reduces its affinity
to be taken up by the
reticuloendothelial system
relative to Qdots coated
with just peptide. The
results for this are shown
in Figure 2 where Qdots
coated with LyP-1 are
compared with Qdots coated
with LyP-1 and PEG.
This result is beneficial in
allowing the drug to stay in
circulation longer and
reducing the dose needed.
One of the problems with
using Qdots is that
fluorescence is not seen
incorporated within the
cells. This is
possible due to the large
size of the Qdot-peptide
conjugate or due to the fact
that the Qdots were not
stable enough to luminesce
within cells and tissue.
There is also a possibility
that the pH of the
microenvironment causes a
quenching of the Qdot
fluorescence.
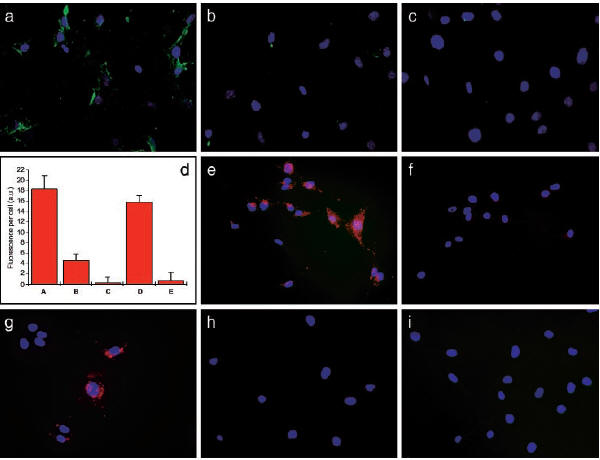
Fig. 1: (a) Qdots
coated with GFE (green) bind
to LE cells (blue) through
the membrane dipeptidase
receptor, but is inhibited
when free GFE (b) or
cilastatin (an inhibitor of
membrane dipeptidase) (c).
(d) shows a bar graph
representation of the amount
of Qdot-GFE incorporation
into LE cells. Column
A is the Qdot-GFE conjugate,
Column B is when free GFE is
added, and Column C is when
cilastatin is added.
Column D shows that binding
to membrane dipeptidase by
Qdot-GFE is not inhibited
when a control peptide is
added. Column E shows
that Qdot-LyP-1 does not
bind to LE cells. (e)
shows that Qdot-F3 bind to
MDA-MB-435 breast carcinoma
cells while it is inhibited
when free F3 is added (f).
(g) shows Qdot-LyP1 binding
to MDA-MB-435 cells.
(h) shows Qdot-GFE does not
bind to MDA-MB-435 cells and
(i) shows Qdot-LyP1 does not
bind to LE cells.
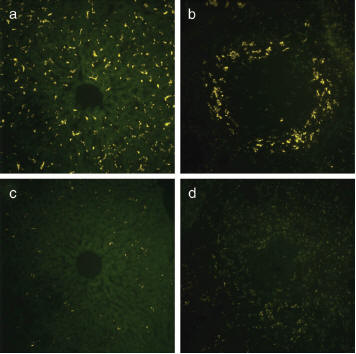
Fig. 2:
Green Qdots coated with
LyP-1 and either with or
without PEG were injected
into the tail vein of a
mouse and the incorporation
into the liver and spleen
were examined by
fluorescence microscopy.
Qdot-LyP-1 is incorporated
into the liver (a) and
spleen (b).
Qdot-LyP-1-PEG shows
significant reduced
incorporation in the liver
(c) and spleen (d).
Y.
Chau, R. Langer, "Antitumor
efficacy of a novel
polymer-peptide-drug
conjugate in human tumor
xenograft models"
Tumors
possess unique
pathophysiology including
highly permeable vasculature
and poor lymphatic drainage.
A high molecular weight
polymer-drug conjugate could
then in theory extravasate
into tumor tissues but not
into normal tissue with low
permeability. Once the
conjugate is inside the
tumor tissue, it cannot
readily exit because of the
poor lymphatics. This
phenomenon is known as
enhanced permeation and
retention (EPR).
Langer's study is designed
to create a drug/polymer
conjugate that targets
releases drug in the
presence of
matrix-metalloproteinase-2
and
matrix-metalloproteinase-9
(MMP-2 & MMP-9,
respectively). The two
enzymes are key in tumor
metastasis and are utilized
for targeted tumor therapy.
Dextran was used as the
polymer in this study due to
its high molecular weight
and its hydrophilic nature
and also due to its
established biocompatibility
and biodegradibility.
The drug chosen was
methotrexate (MTX) which is
a folic acid analog that
inhibits dihydrofolate
reductase and retards the
synthesis of RNA and DNA.
The MTX is linked to the
Dextran by a peptide which
can be cleaved by MMP-2 and
MMP-9 (Figure 1).
Through this, the drug will
be released in the presence
of the tumor and not in a
healthy tissue site.
Three human tumor cell lines
were used and tested for
MMP-2 & MMP-9 expression
(Figure 2). The tumor
lines are HT-1080, U-87, and
RT-112. Tumor lines
HT-1080 and U-87 showed
strong expression of the two
MMPs while RT-112 showed
relatively no expression of
either MMP. In vivo
tests were done in mice
grafted with these tumor
lines. Mice grafted
with HT-1080 or U-87 showed
an inhibition of tumor
growth when the Dextran-MTX
conjugate was administered
relative to the control and
free MTX. Mice that
had the RT-112 line grafted
showed no retardation of
tumor growth when the
conjugate was administered
relative to the control and
free MTX (Figure 3).
This study validates that
the Dextran-MTX conjugate is
an effective drug delivery
device to target tumors that
express MMP-2 and MMP-9,
which are characteristic of
malignant, metastasizing
tumors. Further
studies are being conducted
to determine the severe and
acute drug-related toxicity
of the conjugate. This
study shows that tumor
growth can be stopped but
does not prove that the
tumor can be eliminated
altogether.
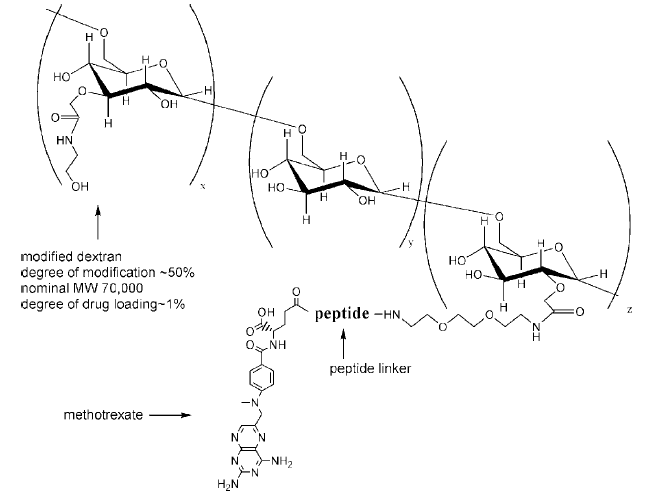
Fig. 1: Chemical
structure of the Dextran-MTX
conjugate. The peptide
linker is designed so that
MMP-2 and MMP-9 cleave the
linker thereby releasing the
MTX in the presence of the
tumor.
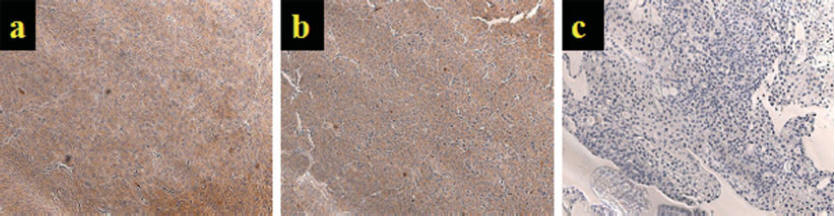

Fig. 2: Top panel is
a staining for MMP-2 and
bottom panel is staining for
MMP-9. Tumor lines
HT-1080 (a), U-87 (b), and
RT-112 (c). In both
panels, a brown indicates a
positive stain for the
respective MMP.
HT-1080 and U-87 show strong
expression of MMP-9 and
sparse expression of MMP-2
while RT-112 shows low
expression levels for both
MMPs. Both panels are
magnified at 20X.
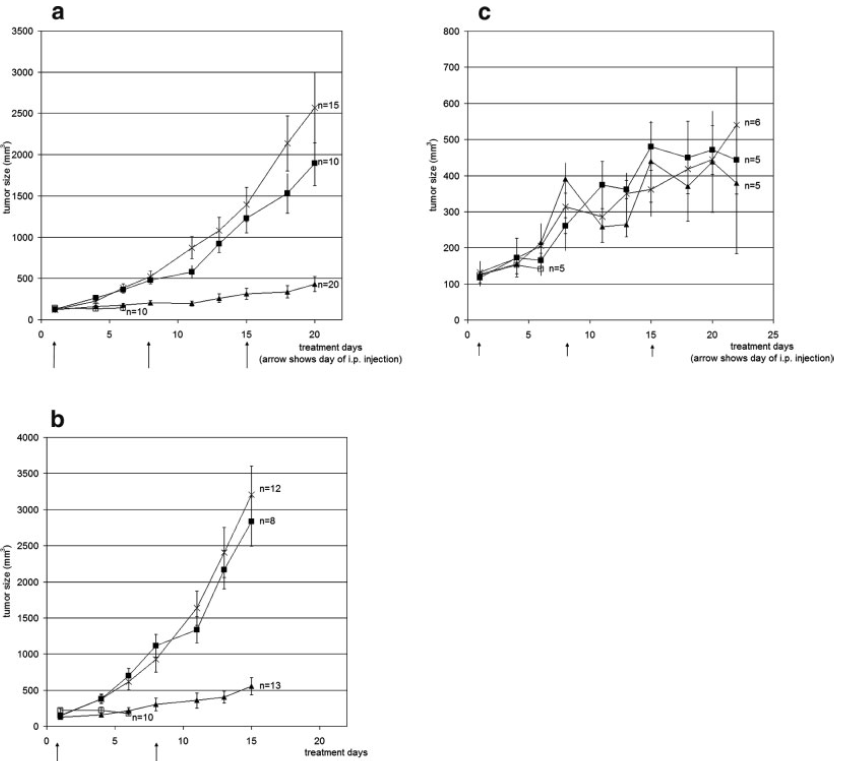
Fig. 3: In vivo tumor
progression of the three tumor lines. (a) HT-1080, (b) U-87, and
(c) RT-112. Control group was treated with PBS (line with X
through it). Free MTX group (line with filled black boxes).
Dextran-MTX group (line with filled triangles). No retardation of
tumor growth in RT-112 line due to lack of MMP-2 and MMP-9 expression
(Figure 2).
|
